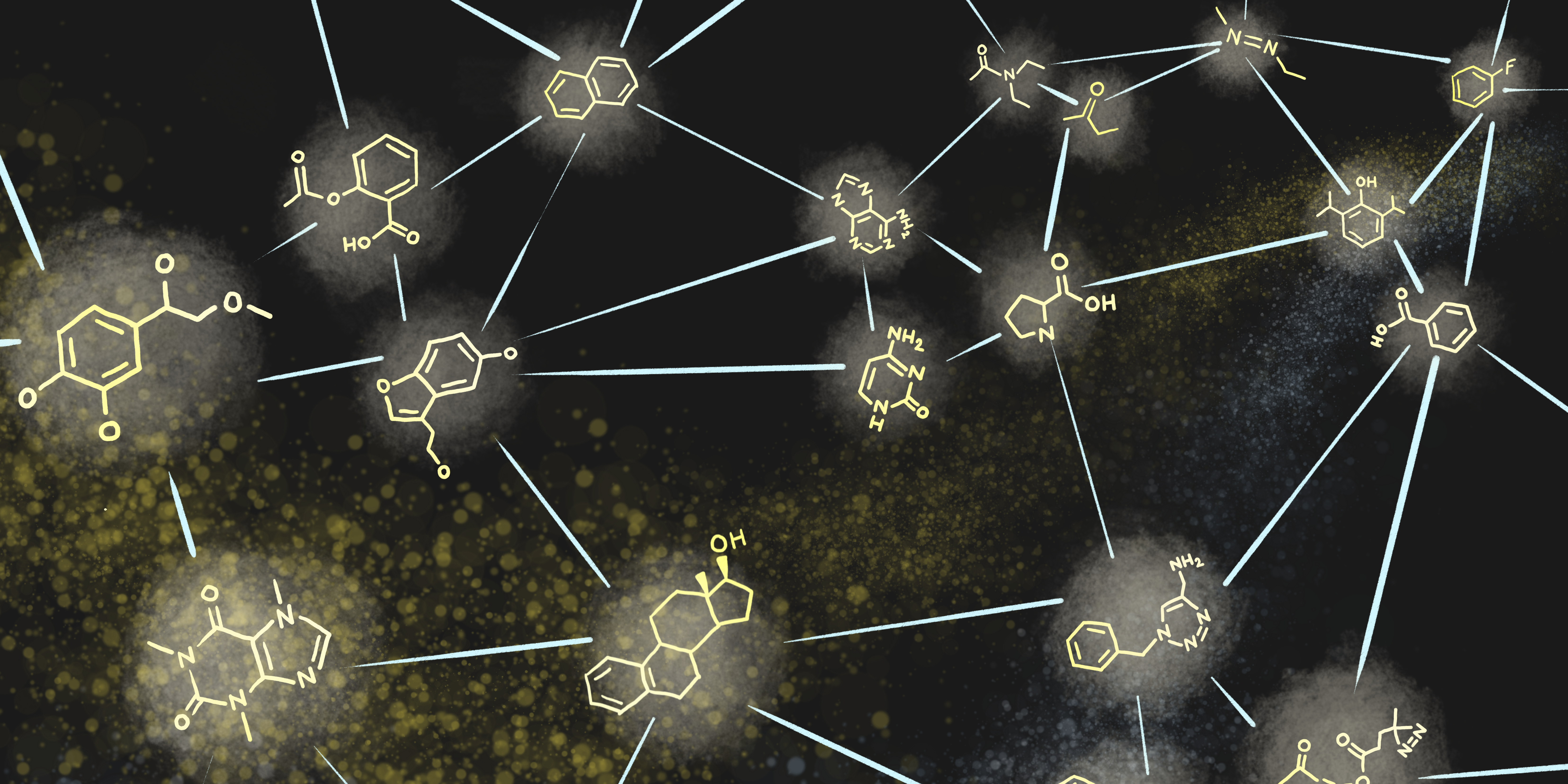
Molecular Assembly is an application of Assembly Theory, which is a novel theoretical
framework developed to characterize the information required to build objects.
Molecular Assembly has applications in
detecting alien life and searching
chemical space for drug candidates. This website allows users to look up the Molecular
Assembly Index (MA) of molecules, learn about how MA is calculated, and how to integrate
this data into their own workflows.
More details on Molecular Assembly and its application can be found in our published research articles.
Contact
For questions please contact Dr Cole Mathis, Keith Y. Patarroyo or Professor Lee Cronin.Citing this work
If you use the data provided here, please cite this work using the following publication:
Identifying molecules as biosignatures with assembly theory and mass spectrometry, Stuart M. Marshall, Cole Mathis, Emma Carrick, Graham Keenan, Geoffrey J. T. Cooper, Heather Graham, Matthew Craven, Piotr S. Gromski, Douglas G. Moore, Sara. I. Walker & Leroy Cronin Nat. Commun., 2021, 12, 3033.
Acknowledgements
Assembly Theory is being developed by Professor Leroy Cronin and Professor Sara Imari Walker thanks to financial support from UK EPSRC, Templeton Foundation and NASA. Stuart M Marshall developed the first algorthmic implementation of Molecular Assembly calculations, with help from Dr. Alon Henson. Dr Yu "Ernest" Liu helped develop additional methods for MA calculations with support from Dr. Doug Moore, Stuart Mashall, and Dr Cole Mathis. This website was built by Dr Cole Mathis and Keith Y. Patarroyo with support from Professor Leroy Cronin, Dr Doug Moore and Stuart Marshall, thanks in part to a template provided by XD-DENG, and support from the NASA Postdoctoral Program.
